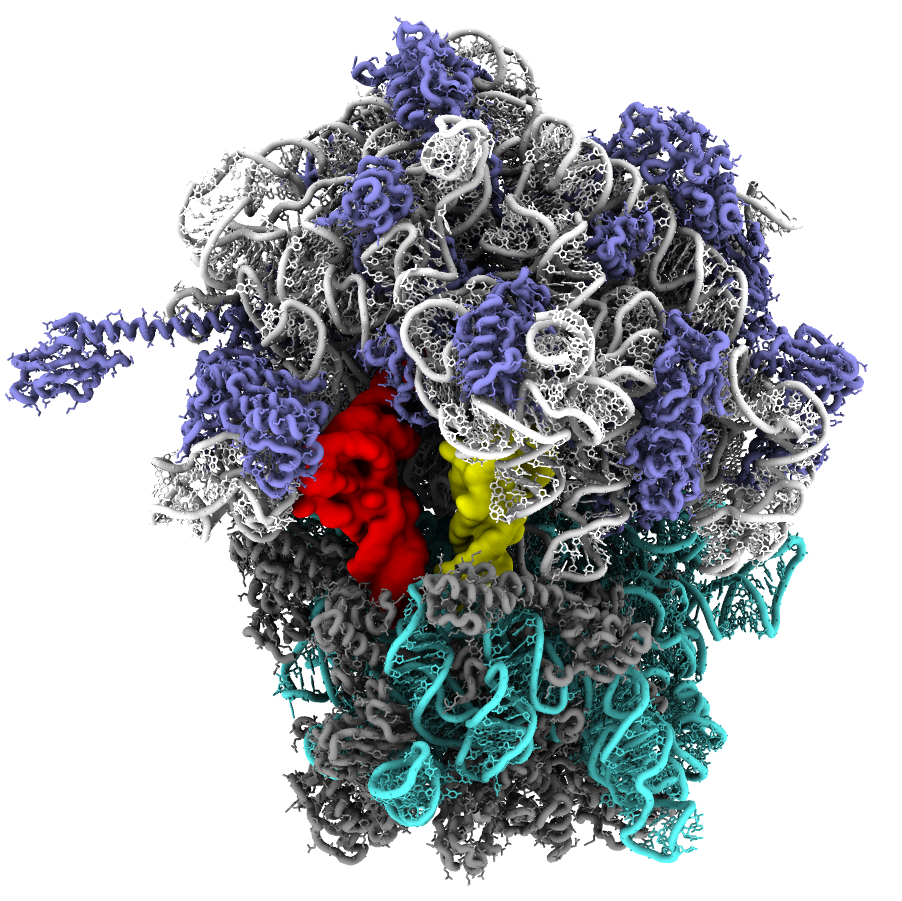
Quantifying Ribosome Energetics
In our group, we use a combination of theoretical concepts and computational tools to understand the relationship between structure, energetics and dynamics in molecular assemblies. In this direction, our most developed line of inquiry has focused on the ribosome, for which we have focused on understanding long-time (ms) and large-scale (1-10nm) conformational dynamics.
The ribosome is a massive molecular assembly that is responsible for converting mRNA sequences into proteins. In order to accomplish this, tRNA molecules (yellow and red in the figure above) must bind the ribosome and transit across the complex, or approximately 25 nm. This involves a number of large-scale rearrangements in the tRNA molecules, as well as in the ribosome.
In our studies of the ribosome, we have used simulations (usually with SMOG models) to describe almost every large-scale rearrangement associated with the elongation cycle. Below are descriptions of each step, in the order in which they occur during elongation.
tRNA Accommodation Accommodation is the process by which a new tRNA molecule binds the ribosome, as it reads a codon in the mRNA. For this step, we have analyzed experimentally-accessible measures of the dynamics, in order to precisely describe the associated free-energy barrier (Noel et al. Biophysical J. 2014). We have also asked how Elongation Factor Tu can affect accommodation, both in terms of entropic and energetic contributions (Noel and Whitford, Nature Comm. 2016). Through a perturbative approach, we also showed how the L11 stalk can sterically confine the tRNA during accommodation, which can facilitate the rearrangement (Yang et al. J Phys Chem B, 2017). More recent efforts have used explicit-solvent simulations to quantify the coordinate-dependent diffusion of tRNA in the ribosome (Yang et al. J Chem Phys, 2019) and we developed an explicit-ion model that allowed us to study how Mg2+ ions can shift the energy landscape of accommodation (Wang et al. J Amer Chem Soc. 2022). For a complete list of studies, see Publications page.
P/E Hybrid State Formation After accommodation, the nascent protein chain is transferred from the P-site tRNA to the A-site tRNA. Then, the P-site tRNA can move towards the E site, a process known as P/E hybrid formation. In our study of this motion, we simulated numerous conformational events and calculated the associated free-energy barrier with our model (Levi et al. Nature Comm. 2020). This revealed how a 6-residue gate-like region is responsible for controlling the kinetics. This region is conserved in bacteria, suggesting that it may represent a novel antibiotic target site.
A/P Hybrid State Formation Next, the A-site tRNA can move towards the P site of the ribosome. In our initial study of this step, we asked whether current single-molecule probes can accurately describe the energetics of this motion (Nguyen and Whitford, J Phys Chem B, 2016). We then built upon this by comparing the free-energy barriers and kinetics for different tRNA species (Nguyen et al. J Phys Chem B, 2017). This showed how the length of the so-called variable loop can shift the landscape and influence the free-energy barrier.
tRNA-mRNA Translocation Once the tRNA molecules have adopted hybrid conformations, the mRNA (bound to tRNA) can move between sites on the ribosome. This step allows the ribosome to ‘reset’ and then bind a new tRNA molecule (accommodation). Using our models, we simulated more than 1000 translocation events (Nguyen and Whitford, Nature Comm. 2016) and showed how the steric composition of the ribosome leads to transient tilting of the small subunit head domain (see video below). More recently, we performed a comprehensive analysis of available experimental measures of head motion (Hassan and Whitford, J Chem Phys B, 2022) and found that current strategies are unable to distinguish between different modes of head rotation. This shows how next-generation single-molecule probes may be able to unambiguously report on the head orientation.
Small Subunit Rotation During hybrid formation and translocation, there is also transient rotation of the ribosomal small subunit. This involves collective rotation of roughly 40,000 non-hydrogen atoms, relative to approximately 100,000 atoms. We have developed and applied a coarse-grained model to simulate this motion in bacteria (Levi et al, Biophysical J 2017; Levi and Whitford, J Phys Chem B, 2019) and we developed a similar all-atom model for studying rotation in a eukaryotic ribosome (Freitas et al. Biophysica, 2021). Recently, we extended this approach to quantify the contribution of ionic interactions to the dynamics of subunit rotation (Wanes et al. PNAS, 2026). Below are videos that show representative trajectories from these studies.
Crystallographic Lattices of Ribosomes In a recent study, we also asked how crystallographic contacts can impact the flexibility of the ribosome (Oliveira et al, Protein Science, 2022). For this, we compared the dynamics of a ribosome in isolation with that of a ribosome in a crystallographic lattice (shown below). While the crystallographic construct is not directly related to biology, this quantitative analysis provides a foundation for gaining insights into dynamics from crystallographic studies.