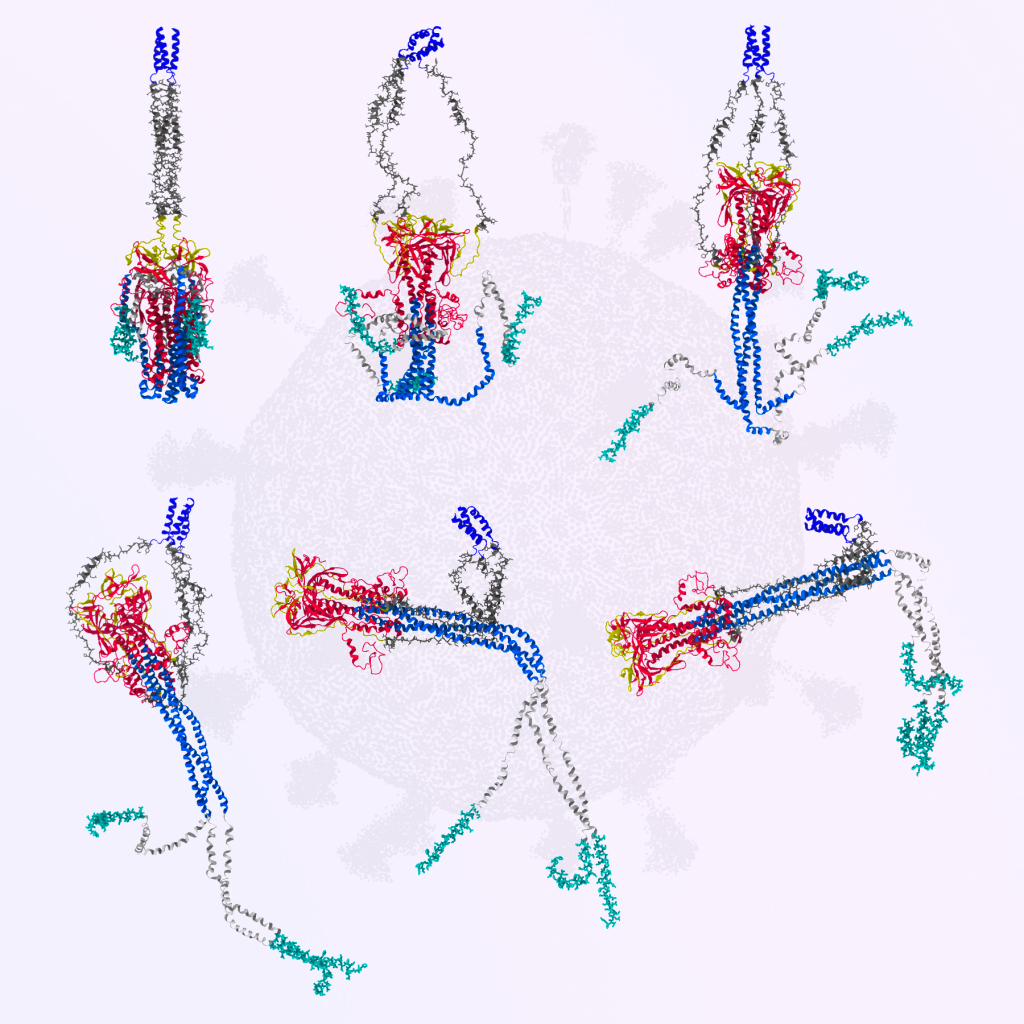
Describing the SARS-CoV-2 Spike protein
Adopting the methods that we have used to study the ribosome, we have more recently moved into the study of the SARS-CoV-2 spike protein. In this line of work, we have focused on understanding how the protein can undero an intricate conformational rearrangement (shown above) that is required for the virus to enter a cell. Through use of our SMOG class of models, we compared the dynamics when the protein is glycosylated and not glycosylated. This comparison showed how the protein gets ‘stuck’ when the glycans are present, where the intermediate allows the protein to more easily bind and infect the host/human cell. The predicted dynamics were originally published in eLife in 2021. Subsequently, we compared our dynamics to cryo-ET reconstructions, which confirmed the mechanistic aspects. Inspired by this, we further asked how a newly-identified antibody can impede this motion and thereby neutralize the virus (See Grunst et al. Science, 2024). For an overview of the simulated dynamics and experimentally-identified states, see the press release videos, below.
Since our simulations provided the first molecular description of the cell-fusion process, they have inspired much discussion in the field. Our simulations were even used to help generate the educational video below.
In parallel, we have also collaborated with experimental groups to develop a novel vaccine delivery platform. By understanding how fragments of the spike protein behave, our calculations have helped guide the identification and implementation of specific epitopes that are particularly effective at eliciting an immune response. In addition, this platform does not require cold storage, which can make it an effective strategy for global distribution of vaccines, including to more remote locations. Our study appeared in PNAS and a visual depiction of the approach is given in the following press release video.